Тема: Алгоритмы анализа белковых последовательностей с использованием внутренних фрагментных ионов
Закажите новую по вашим требованиям
Представленный материал является образцом учебного исследования, примером структуры и содержания учебного исследования по заявленной теме. Размещён исключительно в информационных и ознакомительных целях.
Workspay.ru оказывает информационные услуги по сбору, обработке и структурированию материалов в соответствии с требованиями заказчика.
Размещение материала не означает публикацию произведения впервые и не предполагает передачу исключительных авторских прав третьим лицам.
Материал не предназначен для дословной сдачи в образовательные организации и требует самостоятельной переработки с соблюдением законодательства Российской Федерации об авторском праве и принципов академической добросовестности.
Авторские права на исходные материалы принадлежат их законным правообладателям. В случае возникновения вопросов, связанных с размещённым материалом, просим направить обращение через форму обратной связи.
📋 Содержание
2 Постановка задачи 1
3 Обозначения 2
4 Описание и подготовка данных2
5 Проверка статистических гипотез 2
6 Подход с использованием сырых спектров 3
7 Подход с использованием деконволюцированных спектров 5
7.1 Начальные и конечные ионы 5
7.2 Внутренние ионы 7
8 Подход с использованием лесенок 8
8.1 Определение лесенок 8
8.2 Алгоритм нахождение лесенок 9
8.3 Примеры найденных лесенок 10
8.4 Анализ позиций разломов 12
9 Заключение 12
10 Исходный код решения
📖 Введение
В протеомике масс-спектрометрия используется для идентификации белков и пептидов. Существует два основных подхода, top-down и bottom-up. В случае bottom- up подхода, белок предварительно расщепляется на короткие пептиды, top-down подход использует весь белок целиком.
В работах был представлен алгоритм Twister, решающий задачу de novo секвенирования белков, то есть идентификации последовательности аминокислот в белке на основе набора top-down масс-спектров. Данный алгоритм опирается на тот факт, что фрагментные ионы, соответствующие пикам в масс-спектре, часто образуют последовательности расширяющихся фрагментов с общим концом. Алгоритм в первую очередь опирается на ионы, являющиеся началом или концом всего белка, но в масс-спектрах бывают также и внутренние ионы. Информация об их устройстве и распределении может помочь усовершенствовать алгоритмы de novo секвенирования белков.
В данной работе предполагается проанализировать поведение внутренних фраг- ментных ионов в масс-спектрах.
2 Постановка задачи
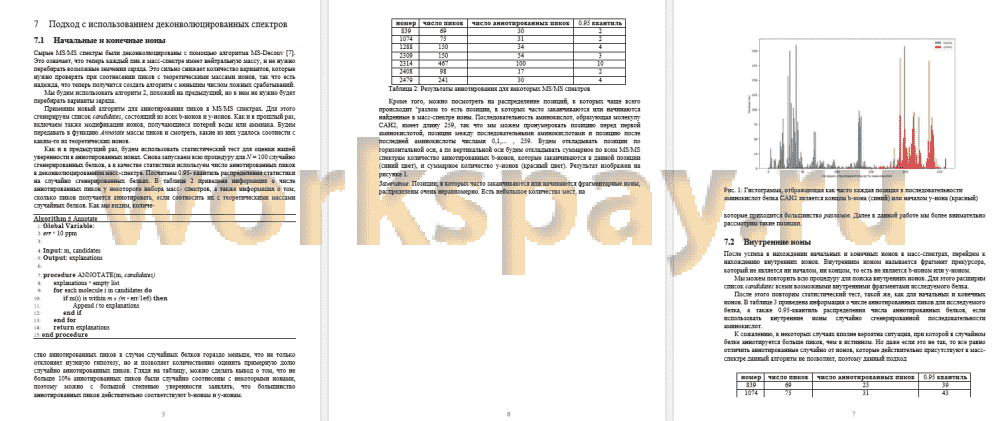
Масс-спектр представляет из себя набор пиков, состоящих из величины отношения массы к заряду и интенсивности. При проведении исследований получается множество масс-спектров, не все из них являются масс-спектром для всего белка, часть из них соответствуют префиксу, суффиксу или инфиксу белка. Часть масс-спектров могут соответствовать посторонним молекулам, случайно попавшим в смесь. Но информация о том, чему соответствует каждый масс-спектр неизвестна и должна быть получена алгоритмическими путями. Задачей является исследование top-down масс- спектров для известного белка CAH2. Для каждого масс-спектра требуется понять, из какого фрагмента белка он был получен, а также разметить пики в этом масс- спектре. Пики могут быть отнесены к начальным, конечным, а также внутренним фрагментам ионов. Кроме того, необходимо проанализировать полученные результаты и попытаться найти закономерности в том, в каких позициях последовательности аминокислот чаще всего начинаются или заканчиваются внутренние ионы.
✅ Заключение
📕 Список литературы
🖼 Скриншоты